From our work on functionally important protein motions, we discovered the mechanism of spatial correlations that lead to highly directed forces to drive biological processes on time scales faster than complete vibrational relaxation into all modes (uniform heating and entropic loss in efficiency). However, what about biological processes known to occur on time scales comparable to decoherence times of transiently occupied vibrational or electronic quantum states. How highly optimized are biological systems? Is it possible that the phase of quantum states is exploited to direct energy (excited states, either vibrational or photoexcited in origin)?
To be more specific, quantum mechanics dictates that all matter has inherent wave properties. On a molecular scale, this property can lead to destructive and constructive interferences that have a pronounced effect on transmission probabilities along reaction coordinates, for example the photo-induced isomerization of the retinal molecule in rhodopsins. Our goal has been to explore whether quantum coherences can persist in nature such that pure quantum or phase related interference effects could play a role in functional responses. The timescales for electronic and nuclear decoherences are on the 10 to 100 fs time and 100 fs to ps time scales, respectively. This time scale is within the domain of barrier crossing dynamics to which proteins/enzymes have evolved to control barrier heights. The time and length scales of the underlying time dependent matter waves are consistent with quantum interference effects. The question reduces to the relevant decoherence timescales in relation to the barrier crossing dynamics leading to energy transduction and functions. Alternatively, one can consider if pure quantum (phase) effects are involved in directing excited state energy as in photosynthetic light harvesting systems. We have investigated several model systems, including dyes in solvents (artificial systems) and bacteriorhodopsin (a natural system) to address this question.
Our initial test system was Rhodamine 101 in methanol, where we were able to control the degree of population transfer from the ground to the excited state using laser pulses with specifically shaped phase and amplitude profiles at weak excitation where the response is linear with respect to the absorbed energy (see Prokhorenko et al J. Chem. Phys. 2005). Using a home-built coherent control setup (and computer-based feedback loop, after the methodology of Judson and Rabitz) we found that the temporal profile of the optimal pulse shape consists of a series of subpulses spaced apart by ~ 150 fs. Further, the optimal spectrum consists of a series of peaks spaced apart by 6.5 nm, corresponding to a frequency of 220 cm-1. This matches the frequency modulation of 150 fs in the population kinetics observed using pump-probe spectroscopy, indicating that even such large systems with many degrees of freedom are sensitive to constructive and destructive interference pathways created by shaped excitation pulses. The current interpretation is that the rhodamine 101 S0-->S1 population transfer can be manipulated by keeping the underlying vibrational states in a coherent superposition during the excitation time period, thus modifying the resonant excitation (via time dependent Franck Condon factors that weight the transition probability). Similarly in bacteriorhodopsin, random fluctuations among the enormous number of degrees of freedom of protein might be expected to cancel any quantum interference effects. However, proteins are highly evolved structures and the question arises whether the phases of the underlying matter waves could play a role or even be manipulated in directing biological processes. To answer this, we have performed an experimental test of the possibility to manipulate the isomerization quantum yield of retinal in bacteriorhodopsin, a key molecule in light harvesting and vision, at very weak excitation levels (relevant to natural biological conditions) using a coherent control approach (see Prokhorenko et al Science 2006, Prokhorenko et al Chem. Phys. 2007). By manipulating the shape (amplitude/spectral components) of the ultrashort laser pulse that jump-starts retinal's isomerization, we were able to enhance or suppress retinal isomerization yield by 20% in either direction. We found the effect depends on the phase of the light field or timing of the various excited nuclear motions – albeit a very small effect of a few percent. The latter effect was consistent with constructive interference effects along the reaction coordinate defined by the protein structure but could not show a decrease in the transition. This experimental observation suggested that the wave properties of matter can play a role in biological processes to the point that they can even be manipulated. This work also led to a very controversial debate in coherent control and measurement for closed and open quantum systems. It has since been shown by measuring quantum yields for photoisomerization by the Mathies group that there is a phase dependence for the initial step, which the work from this group (Johnson et al) showed occurs within a 30 fs component (less than 50 fs), which is faster than electronic decoherence in related molecules. The extremely fast time scale – truly the speed limit for the primary step of vision – enables any phase effect to be observe or rationalized. The effect is still under debate. Even if phase effects are born out, they are not significant enough to be considered an optimized mechanism for biological functions.
This work on coherent control and issue of open and closed quantum systems is all the more remarkable in that this fundamental bit of physics was unearthed using a biological system as the quantum object or test bed.
In parallel to this effort, we have developed new optical methods to perform coherent multidimensional spectroscopies to directly dissect inhomogeneous lineshape and directly determine coherence times scales for electronic and vibrational degrees of freedom. The ultimate goal in this endeavor is to understand how changes in electron distribution affect the interatomic forces, giving rise to the ensuing excited state dynamics and chemical processes. Within the conventional approach using the Born-Oppenheimer (BO) approximation to think about time dependent molecular wavefunctions, the wavefunction is factored into electronic and nuclear wavefunctions. Coherent multidimensional spectroscopy gives information on bright states, those that lead to changes in absorption. Ideally, one would like a direct measure of the nuclear wavefunction to fully understand the problem. As discussed, the group has developed ultrabright electrons sources that literally light up atomic motions and give directly all the nuclear motions – and by the time dependent velocities the very forces involved (see section on Femtosecond Electron Diffraction). The use of all optical methods enable full characterization of the electronic and vibrational population of bright states; whereas electron diffraction allows correlation of all nuclear motions, including dark states, to the evolving electronic or vibrational population dynamics – a complete picture.
With Coherent Multidimensional Spectroscopy and Femtosecond Electron Diffraction, we have fully characterized time dependent molecular wavefunctions. The breakdown in the BO approximation for example occurs during chemical reactions involving conical intersections. At this point, there is strong vibrational nonadiabatic coupling to the electronic states such that we can no longer clearly separate the electronic and nuclear degrees of freedom. This occurrence appears to be more general that previously thought (or would like). The ability to directly observe the far from equilibrium atomic motions in this region provides the missing information needed to fully understand the wavefunction propagation in the barrier crossing region.
This section will give a brief overview of the general principles discovered using coherent multidimensional or 2D Photon Echo Methods.
Life Starts with Water - Dynamic Structure of Liquid H2O
One of the long term objectives of the group was to experimentally determine the many body potential of liquid H2O – the “liquor of life”. This interest is a natural extension of our interest in biological systems and solution phase chemistry. To this end, two new spectroscopic methods have been developed based on diffractive optics. The group along with the Nelson group at MIT can be credited with the development of diffractive optics based nonlinear spectroscopy. This single advance has led to phase stabilized pulse sequences that can be generated from a single optic. This development solved a long standing problem in optical analogues of NMR with respect to generating phase locked pulse sequences (see Cowan et al Chem. Phys. Lett. 2004, Goodno et al J. Opt. Soc. Am. B: Opt. Phys. 1998, Miller et al. Acc. Chem Res. 2010). This development in turn has made it possible to routinely execute heterodyne detection in which the signal to noise is typically increased over a factor of 100 over homodyne or intensity based detection methods. This increased sensitivity and ability to encode even complex beam patterns with the desired phase matching beam geometries using diffractive optics opened up the field of high order nonlinear spectroscopy – namely 6-wave mixing processes. Each different nonlinear interaction probes a different aspect of either electronic or nuclear degrees of freedom that lead to direct determinations of the operating decoherence time scales in the frequency correlations (or memory) of the degree of freedom of interest as well as anharmonic coupling to other degrees of freedom. This information is exactly that needed to build up a first principles understanding of the dynamic structure of liquids and other complex systems. Two different spectral domains have been explored in this context.
The use of 2D-IR spectroscopy gives an unique probe into coupling between vibrational modes and inhomogeneous/homogeneous broadening and dynamics. The OH stretch region of liquid water provides a direct probe of the hydrogen bond network giving rise to water’s unique properties. Unfortunately (or fortunately as discussed below with respect to laser surgical applications), the absorptivity of water is so high in this region that pathlengths on the 100 nm scale are needed to remain in the low OD limit for nonlinear spectroscopy. Prior to the group’s work, it was not possible to study pure H2O. Studies defaulted to isotopic variants with off resonant OD probes of the hydrogen bond structure. The development of diffractive optics and more importantly for this specific problem, the development of nanofluidic cells for this problem made it possible to study the fully resonant network of pure H2O (see Cowan et al Nature 2005, Kraemer et al PNAS 2008). We found the remarkable result that water loses its so-called memory in less than 100 femtoseconds, more than an order of magnitude faster than any other liquid. The degree of hydrogen bonded play an extremely important role. Relatively small changes in lowering the temperature and increasing the degree of hydrogen bonding led to dramatic increases in the coherence time of the OH stretch. The strong dipole-dipole coupling between waters leads to spatial migration of the excitation that is actually faster than the decoherence time. The picture that has emerged is that the OH stretch in pure water is not localized on a single water but is rather spatially delocalized forming a vibrational exciton in which the number of waters coherently involved in the effective vibrational mode(oscillating dipole and induced polarization) depends sensitively on temperature and spatial coherence within the hydrogen bond network. These extraordinarily fast dynamics, spatial coherence, and energy redistribution in water’s hydrogen bond network have profound implications for biological systems. This fast rearrangement of hydrogen bonding provides the correlation in the hydrogen bonding structure of water to coax proteins/biomolecules to find their biologically active conformational states. (Water “wants” to form fully hydrogen bonded networks that have much larger occupied volumes than purely disordered liquid domains. Think ice versus liquid water with respect to density. In this respect, water has spatial correlation that would appear at nano-icebergs expanding against proteins and associated fluctuations, coercing them into minimum volume structures….driving force for protein folding is hydrophobic collapse driven by these forces and spatial correlations in hydrogen bond network of water.)
We now have a completely different model for liquid water from that of a localized basis. It is also clear that the current level of ab initio theory for liquid water is insufficient as the calculations must include >20 waters in an exact manner just to capture the vibrational mode coupling leading to delocalized vibrational excitons and energy redistribution.
In addition to the above formally coherent 4-wave mixing spectroscopy (c3) to study liquid water, the group was the first to implement 6-wave mixing methods to exploit nonresonant Raman scatter from low frequency bath modes to directly probe the highly anharmonic potential of liquids. There was a controversy over cascade artifacts in this form of spectroscopy, which the application of diffractive optics, specifically designed to generate the correct phase matching geometry to remove cascades from the signal field, solved the problem. The overall objective was to use the differential polarizability (spatial dependence) of Raman scattering to directly determine the anharmonicity in the intermolecular potential of the hydrogen bonded network of water. In this regard, water has the smallest polarizability of any liquid such that new laser technology was needed to extract the extremely fast response of water. Six-wave mixing or Two Dimensional Fifth Order Raman Spectroscopy was successfully implemented for CS2 and for the hydrogen bonded liquid formamide. The results from these studies, and accompanying theory showed that liquids are strongly damped on all relevant time scales. The fifth order Raman signal only stressed this feature. The most important work was on the hydrogen bonded network in liquid formamide where the long-predicted bath photon echo was finally observed. This work showed that local correlations could persist for strongly hydrogen bonded liquids such as formamide and reasonable theoretical potentials were found to explain the observed bath memory (See Li et al., J. Chem. Phys. 128, 234507, 2008).
The plan was to extend Fifth Order Raman spectroscopy to liquid water but the laser technology was not in hand at the time (5 fs pulses with 10 - 100 microjoule energies needed) and there were known issues with multiphoton ionization effects). The transition of the group to open new labs in Hamburg Germany as part of founding a new Max Planck Institute offered the opportunity for further advance electron source brightness to probe water in new ways. We have been able to go beyond the nanofluidics developed for our 2D photon echo studies of liquid H2O to open up the use of electron probes (see section on "Nanofabrication and Novel Materials"). With the first generation of such nanofluidic devices, we have been able to directly observe the H-bond in liquid water using in liquid Electron Microscopy ( see de Kock et al JCP 2020 ). Here the power of electron scattering is evident as electron scattering is able to resolve both heavy nuclei and H atoms whereas a combination of neutron and x-ray scattering would be needed to resolve the relevant features. Future work will be directed to observing the H-bond dynamics directly using ultrabright electron sources for imaging (scattering and diffraction).
"Quantum Biology Revisited."
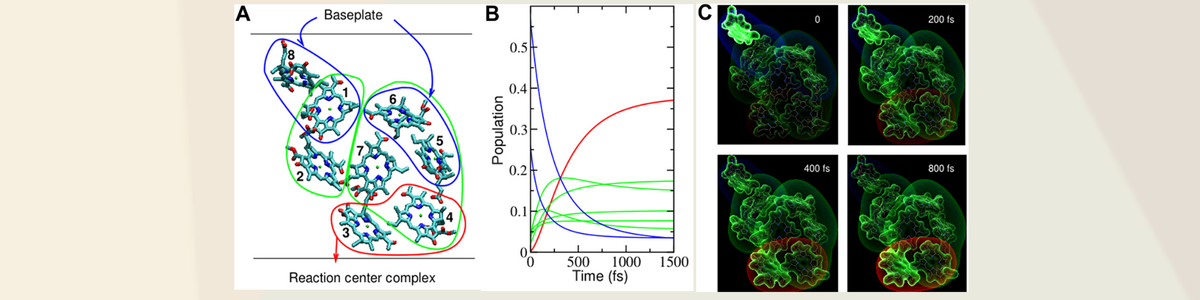
Following up on the discussion of quantum coherence in biological systems, we undertook 2D photon echo studies to look more closely at claims of long-lived coherences in biological systems – notably the assignment of long lived, small amplitude, beats to long lived electronic coherences implying nature had found a way to beat quantum decoherence. This finding was inconsistent with our work on electronic decoherence and understanding of imposed spatial correlations to direct biological processes. This work appeared in PNAS in 2017 (Duan et al) and an authoritative account by 18 authors (Sci. Advances 2020) going back over the details to definitely show that early reports of long lived electronic coherences were misassigned and were really ground state Raman contributions to the 2D spectra. The most important outcome from this work is that it emphasized that Nature understands quantum mechanics very well, including the frailty of quantum phase – especially in biological systems that must exploit strong system-bath coupling. Mother Nature explicitly exploits dissipation and imposes specific structures that create downhill energy gradients that robustly, and highly, direct biological processes (see collective mode coupling molde above in terms of sampling) over all relevant spatial and temporal scales.
This body of work was made possible through a very constructive collaboration with Prof. Michael Thorwart at the University of Hamburg, Hong-Guan Duan as a PhD student (now Professor at Ningbo University), and the Miller group back in in 2017. This collaboration was responsible for definitive experiments on the fundamental issue related to quantum decoherence in biological systems and whether long lived electronic coherence, constructive phase effects unique to quantum aspects of coupled states, could play a role in directing biological processes. The answer was no. Quantum decoherence occurs more than an order of magnitude faster than even the fastest biological processes such as energy transport involved in photosynthesis. However, quantum effects are still everywhere and leave fingerprints in nature if you know where to look. Prof. Thorwart gives a text book discussion of quantum aspects of biological processes and its fundamental basis. This interview is related to a the above mentioned Review published in Science Advances in 2020 to give the present status of experiments and theory with respect to advancing our understanding of quantum effects in biology. See https://advances.sciencemag.org/content/6/14/eaaz4888